
22q: The Missing Piece
22q – A Tiny Piece Leading to a Big Picture
Summary: Donna M. McDonald-McGinn explores 22q11.2 deletion syndrome, the second most common chromosomal abnormality after Down syndrome, which presents with significant, variable, and often overlooked health issues. While commonly involving serious congenital heart disease and developmental delays, early, or prenatal, diagnosis is shown to significantly improve outcomes and reduce intensive care stays for affected infants.

Figure 1
Louis was 12 years old when we published the first iteration of the International 22q11.2 deletion syndrome pediatric healthcare guidelines (10.1542/peds.2011-2903). He was the poster child for 22q (Figure 1), having demonstrated every single associated feature. But you would never know that by speaking with him now. Fifteen years later, Louis remains completely enveloped by his parents, Carol and Louis, his grandmother, sisters, Ashley and Montana, and their now growing family, including brother-in-laws and nieces and nephews whom he sees every single day in his large extended family (below).

Louis (center) with his parents, sisters, and brothers-in-law. (Right) Louis and Carol.
Carol has remained devoted to Louis, along with the international 22q community as the leader of the International 22q11.2 Foundation (22q.org) having just celebrated its 22nd anniversary. I’ve had the pleasure of knowing Louis since infancy, when he first presented with congenital heart disease – specifically tetralogy of Fallot – as I lead the largest center in the world dedicated to providing care for individuals with chromosome 22q11.2 differences at the Children’s Hospital of Philadelphia. I am fortunate to feel like a member of Louis’ extended family, having joined Carol and a handful of other parents in establishing the Foundation all those years ago and while leading multiple rhyming events – think 22q and Boo, 22k for 22q, 2.2 for 22q, and 22q at the Zoo – Worldwide Awareness Day (below). Like Carol, many of the Founders remain committed to supporting the Foundation to this day, even though their children are now also adults, because the mission is so important.

Louis with his mother Carol and the author, Donna McDonald-McGinn at 22q at the Zoo.

Louis has moved into adulthood but remains active in the Special Olympics, participating in every activity imaginable from basketball and swimming to track and field and golf. He's gone to national and international competitions, and his family has always been there to rally behind him. Louis also enjoys weekly art classes where he shines, and like his mother he has begun giving back by creating greeting cards for the Foundation (right).
Louis has the second most common chromosomal abnormality after Down syndrome, which is also the most common cause of syndromic palatal anomalies and schizophrenia, and the second most common cause of congenital heart disease and developmental delay after Down syndrome. But despite a prevalence of 1 in 2148 livebirths, which is one of the more common rare conditions, 22q is likely the most frequent condition you’ve never heard of. So, what exactly is the 22q11.2 deletion?

Well, before discussing chromosomes and genes, we need to back up and think about cells. All living things are made up of cells including a leaf, where the cells are visible as boxes (Figure 5, left). Our bodies are made of different types of cells. We all have blood, brain and heart cells, as well as many other types of cells. In each cell, genetic information is stored in a chemical called DNA, which is packaged neatly into chromosomes. Chromosomes are located within the cell and are visible under the microscope where we expect to find 46, including 22 pairs numbered from the largest (number 1) to the smallest (number 22) plus the sex chromosomes (X and Y). When conceived, we receive one of each chromosome from our parents. Chromosomes have a top and a bottom half where the top half is always smaller. As chromosomes were first described by the French, the top half is referred to as the “petite arm” or p arm for short
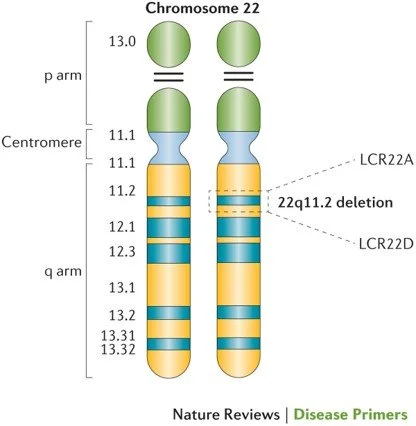
and the bottom half is the q arm, as q follows p in the alphabet. Chromosomes are further defined by a numbering system that names the stripes (or bands) on the chromosome when they are stretched out like an accordion. Those numbers create a universal language, so everyone knows exactly which area of a chromosome is involved. Chromosomes bands are like the box cars on a train - if a boxcar is missing or extra, we see it with our eyes and if a band is missing or extra, we generally see it under the microscope. The genes are the packages inside and they set up a blueprint for how the body is formed - our hair color and eye color, how tall we are, etc. The chromosome 22q11.2 deletion involves missing genes. Nothing the parents did or did not do caused
the deletion to occur. It happens most often spontaneously due to the structure of the chromosome. People with 22q, like Louis, are missing about 50 genes in the 22q11.2 region (Figure 6, right). Once the deletion is present, it is missing in every cell of the body. Currently there is no way to replace the missing genes.
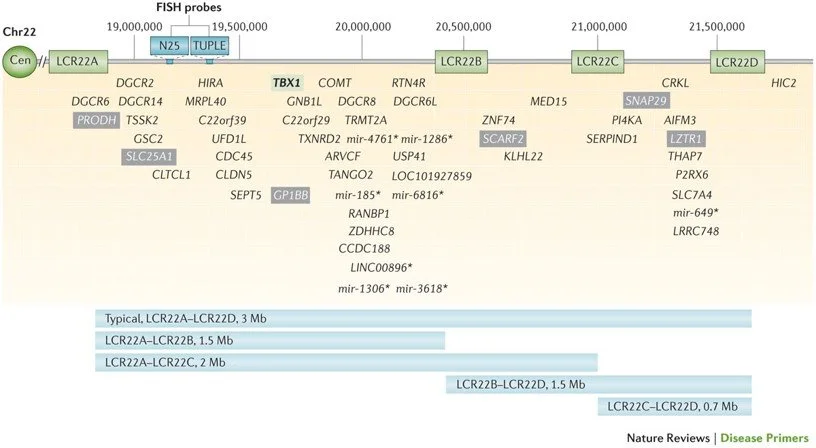
Figure 7
Loss of genes within the 22q11.2 region (Figure 7) affects cells in the developing embryo frequently causing structural abnormalities of the heart, absence or underdevelopment of the thymus gland leading to immunodeficiency, and underdevelopment of the parathyroid glands resulting in low calcium which can cause seizures and other symptoms such as muscle twitching, feeding difficulty and noisy breathing. Louis has had all of these medical issues. But 22q can cause many other problems including abnormalities of the palate affecting speech, significant feeding and swallowing problems – often the most difficult problem for new parents to manage as they often feel like failures when they can’t feed their newborn, renal abnormalities including missing or cystic kidneys, bony problems including scoliosis (curvature of the spine) sometimes requiring a placement of a growing rod, as it did in Louis resulting in multiple spinal surgical procedures, chronic ear and sinus infections, hearing loss, problems with low platelets (the cells that help us to stop bleeding), thyroid abnormalities, eye problems, speech and language delay, differences in learning style – particularly with mathematics, ADHD, autism, anxiety, movement disorders such as early Parkinson’s disease, and psychiatric illness including schizophrenia (Figure 8).
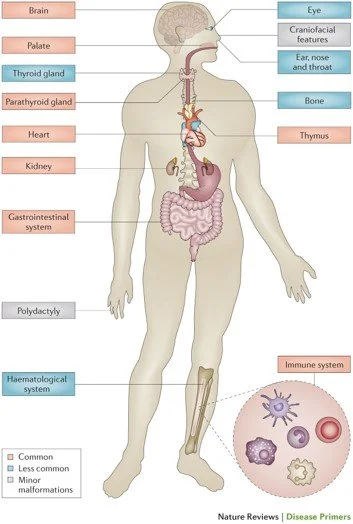
Now despite this very long list of findings, only 4% of our pediatric patients have succumbed to associated problems, most often related to severe congenital heart disease, with a median age at death of 5 months. So, in general, people with 22q grow into adulthood. Moreover, 22q is variable and some individuals have very few of these problems. In fact, although 22q is not usually running in the family (but once present there is a 50% chance of passing it on to a child), many adults have come to attention only following the diagnosis in their child born with congenital heart disease. This variability explains why many families suffer, like so many with a rare disease, from a protracted diagnostic odyssey. For example, a five-year-old was evaluated by a whooping 27 subspecialists, including three geneticists, without a unifying diagnosis, but his parents were relentless in their search for a unifying diagnosis to explain his failure to thrive,
reflux, chronic infection, and delayed developmental milestones, particularly speech. Following the diagnosis of 22q, he went on to benefit from a palate repair to improve his hypernasal speech, growth hormone therapy, and academic support, but his family still bemoans the fact that he may have had associated treatable conditions, like low calcium in the newborn period, that went undiagnosed and may have impacted his long-term outcome.
So, what is the benefit of early diagnosis? We recently examined outcomes in 135 patients with 22q and interrupted aortic arch (IAA) diagnosed before and after birth hospital discharge. We found, with advancements in fetal imaging and non-invasive prenatal screening (examining the fetal DNA by way of a blood test in the mother), prenatal diagnostic rates have improved over time, from 7% in the 1980’s to 75% currently. Importantly, infants with 22q were more likely to deliver in a hospital with a higher-level NICU – if diagnosed prenatally. Moreover, the mean Intensive Care Unit stay for infants receiving a 22q diagnosis prior to neonatal hospital discharge v. post discharge was significantly shorter (18.5 vs 33.1 days). Concurrently we found that 14 children inherited the deletion from a parent (10%), but only 3 parents were aware that they had the deletion before the diagnosis in their child. Thus, we strongly believe that children with 22q benefit from early ascertainment - prenatally wherever possible, as we have demonstrated that prenatal or neonatal diagnosis of 22q, prior to the child being discharged from the newborn nursery, in children with one of the most serious heart lesions, IAA, can decrease associated medical complications and even death, concurrently reducing overall medical costs.
This data demonstrates the need for non-invasive prenatal screening for 22q, which neither the American College of Obstetrics and Gynecology nor the Society for Maternal Fetal Medicine have yet supported. Likewise, it establishes the need for newborn screening for 22q, which has been available since 2012, but no government has yet adopted it. Fortunately, many children are coming to attention via newborn screening for Severe Combined Immunodeficiency (SCID), as children with SCID and those with 22q have similar problems with low T cells, including neonates with unrecognized low calcium requiring treatment who only come to attention via newborn screening for SCID. It is notable that more children with 22q are coming to attention via newborn screening for SCID than children with SCID. But children with 22q without low T cells may continue to go undiagnosed, some even into adulthood. This includes children with IAA, a critical heart defect, who may not have an audible murmur at birth and who may suffer a catastrophic event at home potentially leading to life changing complications such as a stroke. Conversely, early diagnosis results in early treatment – for the heart, the immune system, the endocrine system, etc. all of which in turn help to protect the brain from secondary insults.
Now that we know what we know, what do we do? Well, we follow the fetal, pediatric and adult healthcare recommendations developed by the international 22q11.2 scientific community (22qsociety.org) that emphasizes treating what is treatable (10.1016/j.gim.2022.11.012) – including the brain. Providing recommendations for periodic assessments and management by age and at diagnosis including important do’s and do not’s such as “do investigate feeding and swallowing issues as soon as they present” and “do not assume that feeding difficulty is related to congenital heart disease” and “do recognize that symptoms change over time and family members/caregiver are essential members of the team” and “do not overwhelm families with a list of nonactionable associated features or exclude family members from participating in care discussions.” These recommendations also underscore the fact that transition to adulthood requires a dedicated approach with individualized, multidisciplinary, coordinated care. Recognizing that issues are chronic - requiring longitudinal management, and that the multi-system nature of the condition and potential side effects of treatment demand attention by all clinicians – primary care providers and specialists alike. Requiring an overall manager with familiarity with 22q.
We have only recently moved on from February, which is congenital heart disease awareness month. So, it’s important for everyone to know that 22q is one of the most common causes of congenital heart disease. These include complicated differences such as IAA, truncus arteriosus, and tetralogy of Fallot (like Louis) with and without pulmonary atresia or major aortopulmonary collateral arteries (MAPCAs). But 22q is also a common cause of ventricular septal defects (VSD), atrial septal defects (ASD), bicuspid aortic valve, and abnormalities of the aortic arch, including right aortic arch, double aortic arch, vascular ring, and aberrant subclavian artery. Thus, in February and March, as in all months, it is important to know about 22q because the earlier we make a diagnosis the sooner we can treat associated problems while concurrently helping individuals with 22q, like Louis, and their families.
To learn more about 22q visit:
About the Author

Donna McDonald-McGinn, MS, LCGC, is the Chief of the Section of Genetic Counseling, Director of the 22q and You Center and Senior Principal Scientist at Children’s Hospital of Philadelphia. Areas of expertise: Genetic counseling, Genetics and 22q11.2
References:
Blagowidow N, Nowakowska B, Schindewolf E, Grati FR, Putotto C, Breckpot J, Swillen A, Crowley TB, Loo JCY, Lairson LA, Óskarsdóttir S, Boot E, Garcia-Minaur S, Cristina Digilio M, Marino B, Coleman B, Moldenhauer JS, Bassett AS, McDonald-McGinn DM. Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions. Genes (Basel). 2023 Jan 6;14(1):160. doi: 10.3390/genes14010160. PMID: 36672900; PMCID: PMC9858737.
Boot E, Óskarsdóttir S, Loo JCY, Crowley TB, Orchanian-Cheff A, Andrade DM, Arganbright JM, Castelein RM, Cserti-Gazdewich C, de Reuver S, Fiksinski AM, Klingberg G, Lang AE, Mascarenhas MR, Moss EM, Nowakowska BA, Oechslin E, Palmer L, Repetto GM, Reyes NGD, Schneider M, Silversides C, Sullivan KE, Swillen A, van Amelsvoort TAMJ, Van Batavia JP, Vingerhoets C, McDonald-McGinn DM, Bassett AS. Updated clinical practice recommendations for managing adults with 22q11.2 deletion syndrome. Genet Med. 2023 Mar;25(3):100344. doi: 10.1016/j.gim.2022.11.012. Epub 2023 Feb 2. PMID: 36729052.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015 Nov 19;1:15071. doi: 10.1038/nrdp.2015.71. PMID: 27189754; PMCID: PMC4900471.
McDonald-McGinn DM. 22q11.2 deletion syndrome: A tiny piece leading to a big picture. Am J Med Genet A. 2018 Oct;176(10):2055-2057. doi: 10.1002/ajmg.a.40653. PMID: 30380195; PMCID: PMC6472263.
McDonald-McGinn, DM. The Chromosome 22q11.2 Deletion Syndrome - A Multidisciplinary Approach to Diagnosis and Treatment. 1st Edition - August 24, 2022; Academic Press; Paperback ISBN: 9780128160473; eBook ISBN: 9780128160480
Óskarsdóttir S, Boot E, Crowley TB, Loo JCY, Arganbright JM, Armando M, Baylis AL, Breetvelt EJ, Castelein RM, Chadehumbe M, Cielo CM, de Reuver S, Eliez S, Fiksinski AM, Forbes BJ, Gallagher E, Hopkins SE, Jackson OA, Levitz-Katz L, Klingberg G, Lambert MP, Marino B, Mascarenhas MR, Moldenhauer J, Moss EM, Nowakowska BA, Orchanian-Cheff A, Putotto C, Repetto GM, Schindewolf E, Schneider M, Solot CB, Sullivan KE, Swillen A, Unolt M, Van Batavia JP, Vingerhoets C, Vorstman J, Bassett AS, McDonald-McGinn DM. Updated clinical practice recommendations for managing children with 22q11.2 deletion syndrome. Genet Med. 2023 Mar;25(3):100338. doi: 10.1016/j.gim.2022.11.006. Epub 2023 Feb 2. PMID: 36729053.
Ron HA, Crowley TB, Liu Y, Unolt M, Schindewolf E, Moldenhauer J, Rychik J, Goldmuntz E, Emanuel BS, Ryba D, Gaynor JW, Zackai EH, Hakonarson H, McDonald-McGinn DM. Improved Outcomes in Patients with 22q11.2 Deletion Syndrome and Diagnosis of Interrupted Aortic Arch Prior to Birth Hospital Discharge, a Retrospective Study. Genes (Basel). 2022 Dec 24;14(1):62. doi: 10.3390/genes14010062. PMID: 36672801; PMCID: PMC9859187.